








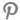





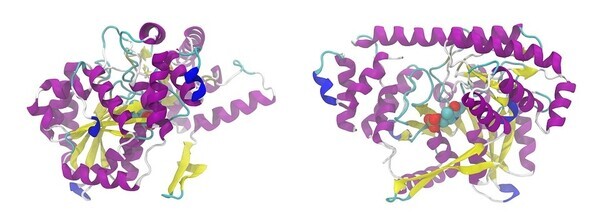
2020년, 런던 스타트업 딥마인드(DeepMind)가 단백질 구조를 예측하는 인공지능(AI) 프로그램 알파폴드(AlphaFold)를 개발했다. 알파폴드는 공개와 동시에 생물학계 난제를 풀 것이라는 기대를 한 몸에 받았다. 최근, 메타가 알파폴드와 같은 AI 단백질 예측 구조 프로그램인 ‘ESM폴드(ESMFold)’를 선보였다.
해외 과학 전문 매체 네이처는 알렉산더 라이브스(Alexander Rives) 메타AI 단백질팀 연구 국장을 인용, 메타 연구팀은 각각 문자로 표현할 수 있는 아미노산 사슬 20종으로 표현할 수 있는 단백질 시퀀스를 알고리즘에 주입했다고 설명했다. 이후 알고리즘의 네트워크는 아미노산의 일부가 가려진 단백질 자동 완성 방법을 학습했다.
라이브스 국장은 단백질 형태 정보를 보유한 단백질 시퀀스를 사용한 뒤 이미 알려진 단백질 구조와 시퀀스 간의 관계 심층 분석 정보를 이용하여 단백질 시퀀스로 예측할 수 있는 구조를 생성하도록 훈련했다.
이후 연구팀은 토양과 해수, 인간의 내장, 피부 및 기타 미생물 서식지를 포함한 환경 원천에서 대량 배열된 유전자학 DNA 데이터베이스에 ESM폴드를 적용해보았다. 이 과정에서 단 2주간 총 6억 1,700만 개 이상의 단백질 구조를 예측했다.
ESM폴드의 단백질 구조 예측 속도는 알파폴드보다 60배 더 빠르다. 그러나 ESM폴드는 아직 알파폴드보다 정확도가 낮은 편이다.
다만, ESM폴드가 예측한 단백질 구조 중 1/3 이상은 질적으로 완성도가 높아, 전체적인 단백질 모양이 정확하다는 확신을 가질 수 있었다. 간혹 더 미세한 원자 수준의 세부 사항을 구별할 수도 있었다.
하버드대학교 진화생물학자인 세르게이 오브친니코프(Sergey Ovchinnikov) 박사는 ESM폴드가 낮은 신뢰도를 기반으로 한 단백질 구조 수억 개를 예측한 방법을 궁금해한다. 오브친니코프 박사는 일부는 적어도 분리된 정의된 구조가 없을 수 있는 반면, 다른 일부는 단백질 코딩 물질로 잘못 알려진 비코딩 DNA일 수 있다고 언급했다.
반면, 독일 뮌헨공과대학교 컴퓨터 생물학자인 버크하드 로스트(Burkhard Rost)는 ESM폴드의 단백질 예측 구조 생성 속도가 빠르다는 점에 감탄했다. 다만, 균유전체학 데이터베이스 사용 시 알파폴드의 예측 결과보다 더 우수하다고 평가할 수 있을지는 더 지켜봐야 한다고 덧붙였다.
[저작권자ⓒ CWN(CHANGE WITH NEWS). 무단전재-재배포 금지]










![[김대선 칼럼] 생(生)의 찬란한 동행과 사(死)의 품격 있는 갈무리](/news/data/2025/12/31/p1065594030678023_224_h.png)


